

Want to share your content on R-bloggers? click here if you have a blog, or here if you don’t.
In our previous post, Examining Meta Analysis, we contrasted a frequentist version of a meta analysis conducted with R’s meta package with a Bayesian meta analysis done mostly in stan using the rstan package as a front end. We did this to hint at the difference between working within the restricted confines of a traditional frequentist framework and the amazing freedom to set up and solve complex probabilistic models using a modern Bayesian engine like stan. However, we fully acknowledge the cognitive burden of learning a completely new language and, at the same time, also learning Bayesian methods.
In this post, we will ease your anxiety by pointing to a middle way by using the well-established and powerful package brms^1 to formulate stan models.
Set Up
First, we set up our environment and read in the data on the effect of Amlodipine on angina patients which can be found in the `meta’ package.
Show the code
library(tidyverse) library(brms) library(tidybayes) library(ggdist)
nE is the number of subjects in the treatment arm for each Protocol, meanE is the sample treatment mean for each protocol, varE is the observed sample variance for each protocol, and nC, meanC, and varC are the corresponding statistics for the control arm.
Show the code
angina <- read_csv(file = "Amlodipine.csv", col_types = c("c",rep(c("i","d","d"),2)))
angina
# A tibble: 8 × 7 Protocol nE meanE varE nC meanC varC1 154 46 0.232 0.225 48 -0.0027 0.0007 2 156 30 0.281 0.144 26 0.027 0.114 3 157 75 0.189 0.198 72 0.0443 0.497 4 162 12 0.093 0.139 12 0.228 0.0488 5 163 32 0.162 0.0961 34 0.0056 0.0955 6 166 31 0.184 0.125 31 0.0943 0.173 7 303 27 0.661 0.706 27 -0.0057 0.989 8 306 46 0.137 0.121 47 -0.0057 0.129
Review The Model
As in the previous post, we will measure the effect of the amlodipine treatment by looking at the difference in the observed means from the two arms of the study. Our model can be expressed as :
![]()
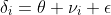
where for each study, i, ![]()
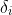
![]()
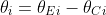
![]()
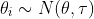
![]()
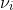
![]()
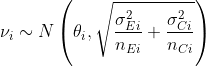
and ![]()
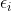
![]()
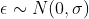
The data include ![]()
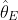
![]()
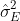
With this, we have that ![]()
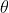
![]()
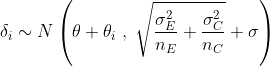
brms Syntax
Although it is much simpler than using stan directly, brms is not without its own cognitive load. Using any complex package correctly and with confidence requires time and effort to understand how things work. At a minimum, it is necessary to fully comprehend the function signature and all of the options implicitly coded therein.
A good bit of the cognitive load associated with brms is associated with the formula interface, which it adopts form the lme4 package for formulating and solving frequentist mixed-effects models. brms builds on this syntax to allow formulating expressions to set up complex, multilevel models.
The general formula argument^2 is structured as response | aterms ~ pterms + (gterms | group). Everything on the right side of ∼ specifies predictors. + is used to separate different effects from each other. Note: the formula syntax is a very compressed notation, so even though it is in itself concise, explanations of it tend to be long. We very much recommend reading the package documentation.
aterms
The aterms are optional terms that provide special information about the response variable. Especially helpful for meta-analysis, the term se specifies the standard errors of the response variable when response is a treatment effect. The pdf states:
Suppose that the variable yi contains the effect sizes from the studies and sei the corresponding standard errors. Then, fixed and random effects meta-analyses can be conducted using the formulas yi | se(sei) ~ 1 and yi | se(sei) ~ 1 + (1 | study), respectively, where study is a variable uniquely identifying every study. … By default, the standard errors replace the parameter sigma. To model sigma in addition to the known standard errors, set argument sigma in function se to TRUE, for instance, yi | se(sei, sigma = TRUE) ~ 1.
pterms
pterms are population-level terms. Everything on the right side of formula that is not recognized as part of a group-level term is treated as a population-level effect.
gterms
gterms are group-level terms that are specified as (coefs | group) where coefs contains one or more variables whose effects are assumed to vary with the levels of the grouping factor. For example, if both a group intercept and subject age vary by group, the group effects would be specified by (1 + age | group). Note that it is possible to specify multiple grouping factors, each with multiple group-level coefficients.
Set up the model using the brms package
First, we read in the data using dplyr , add delta_i the difference in means and its standard error, se_di, rename Protocol to study for convenience, and drop the variables we no longer need. Note: we will refer to Protocol as study from now on.
Show the code
df <- angina %>% mutate(delta_i = (meanE - meanC), se_di = sqrt(varE/nE + varC/nC)) %>%
rename(study = Protocol) %>%
select(study, delta_i, se_di, nE,nC)
head(df)
# A tibble: 6 × 5 study delta_i se_di nE nC1 154 0.234 0.0701 46 48 2 156 0.254 0.0958 30 26 3 157 0.145 0.0977 75 72 4 162 -0.135 0.125 12 12 5 163 0.157 0.0762 32 34 6 166 0.0894 0.0980 31 31
Fit the model
Note that we are using the aterm se to inform the brm() function about varE and varC. The sigma = TRUE flag indicates that the residual standard deviation parameter sigma should be included in addition to the known measurement error. See the epsilon term in the model equations above. prior is an alias for the set_prior() function^2.
The last two lines in the call to brm() specify that the model is to be fitted using 4 chains, each with 2000 iterations of which the first 1000 are warm up to calibrate the sampler. This provides a total of 4000 posterior samples.
Show the code
## Random effects meta-analysis
fit_brms <- brm(
# specify random standard error effect size by group. See p43 of pdf
# sigma = TRUS means estimate epsilon variation in addition to using standard error estimates from data
delta_i | se(se_di, sigma = TRUE) ~ 1 + (1 | study),
data = df,
# set priors in stan language
prior = c(prior(normal(0, 1), class = Intercept),
prior(normal(0, 1), class = sd, group = study)),
iter = 2000, warmup = 1000, cores = 4, chains = 4, seed = 14,
control = list(adapt_delta = 0.95))
# Save an object to a file
saveRDS(fit_brms, file = "fit_brms.rds")
The model fit function shows the great advantage brms achieves by combining lme4s concise formula notation with the exact solutions provided by stan. Working directly in stan does provide great freedom, but freedom comes at the cost of easily specifying models that might not make sense. The brms notation helps guide users in specifying meaningful models.
Note that in most cases, it is not necessary to specify Normal or t-distribution priors. The software will get it right. We included the prior statement to show how it works. Also note that in order to save processing time when running on the blog site, we read in the model fit object from the RDS file which we wrote above.
Here are the results of the model fit.
Show the code
# Restore the object fit_brms <- readRDS(file = "fit_brms.rds") summary(fit_brms)
Family: gaussian
Links: mu = identity; sigma = identity
Formula: delta_i | se(se_di, sigma = TRUE) ~ 1 + (1 | study)
Data: df (Number of observations: 8)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Multilevel Hyperparameters:
~study (Number of levels: 8)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 0.10 0.09 0.00 0.33 1.00 1505 2108
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept 0.16 0.08 0.01 0.34 1.00 2033 1610
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 0.10 0.09 0.00 0.33 1.00 2129 1798
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
The first section of the summary gives some basic information about the model specification. Following this are the statistics for each of the estimated parameters. We see that they are in substantial agreement with the results obtained in our previous post, even though some of the details of the model specification are slightly different than the previous model fits. The payoff is the Intercept, whose value is 0.16.
For each estimated parameter Estimate and Est.Error provide the mean and the standard deviation of the posterior distribution. l-95% CI and u-95% CI provide the lower and upper bounds on the 95% credible intervals. Rhat = 1 for each parameter indicates that the Markov chains have converged. An Rhat considerably greater than 1 (i.e., > 1.1) would mean that the chains have not yet converged, and it would be necessary to run more iterations or perhaps set stronger priors. (See Gelman and Rubin) Bulk_ESS and Tail_ESS are diagnostics for the sampling efficiency in the bulk and tail of the posterior distribution, respectively. See the stan documentation for details and interpretation.
The brms package also provides a plot method that provides posterior density plots and trace plots of the MCMC draws for the estimated statistics. Note that b_Intercept is the internal stan name for the population intercept.
Show the code
plot(fit_brms)

Plots for analysis
Now we will prepare a Bayesian version of a forest plot with the help of the spread_draws() function in the tidybayes package that collects information from the fit_brms object and returns it as columns in a data frame. This first code block creates two data frames out_indiv adds the population to the study specific intercept for each draw, while out_theta sets up the data to compute the mean value of population intercept. These data frames are combined into out_all, which is ready for plotting. The mean_qi() function from tidybayes translates draws from distributions in the grouped data frame into point and interval summaries.
Show the code
# Prepare data frame for plotting
out_indiv <- spread_draws(fit_brms, r_study[study,term], b_Intercept) %>%
mutate(Intercept = r_study + b_Intercept) %>%
mutate(study = as.character(study)) %>%
select(study,term,Intercept)
out_theta <- spread_draws(fit_brms, r_study[study,term], b_Intercept) %>%
mutate(study = "theta") %>%
mutate(Intercept = b_Intercept) %>%
select(study,term,Intercept)
out_all <- bind_rows(out_indiv, out_theta) %>% mutate(study = factor(study)) %>%
ungroup() %>%
mutate(study = str_replace_all(study, "\\.", " "))
# Data frame of summary numbers
out_all_sum <- group_by(out_all, study) %>% mean_qi(Intercept)
This code stacks and plots the graphs.
Show the code
# Draw plot
out_all %>%
ggplot(aes(Intercept,study)) +
# Zero!
geom_vline(xintercept = 0, linewidth = .25, lty = 2) +
stat_halfeye(.width = c(.8, .95), fill = "dodgerblue") +
# Add text labels
geom_text(
data = mutate_if(out_all_sum, is.numeric, round, 2),
aes(label = str_glue("{Intercept} [{.lower}, {.upper}]"), x = 0.75),
hjust = "inward"
) +
# Observed as empty points
geom_point(
data = df %>% mutate(study = str_replace_all(study, "\\.", " ")),
aes(x=delta_i), position = position_nudge(y = -.2), shape = 1
)

Stan Code
Finally, we extract the stan code from the fit_brms object. This is an extremely useful feature for checking the model, checking that your brms code is doing what you think it is, and maybe for learning stan.
Show the code
stancode(fit_brms)
“stan code”
Show the code
stancode(fit_brms)
// generated with brms 2.21.0
functions {
}
data {
int N; // total number of observations
vector[N] Y; // response variable
vector[N] se; // known sampling error
// data for group-level effects of ID 1
int N_1; // number of grouping levels
int M_1; // number of coefficients per level
array[N] int J_1; // grouping indicator per observation
// group-level predictor values
vector[N] Z_1_1;
int prior_only; // should the likelihood be ignored?
}
transformed data {
vector[N] se2 = square(se);
}
parameters {
real Intercept; // temporary intercept for centered predictors
real sigma; // dispersion parameter
vector[M_1] sd_1; // group-level standard deviations
array[M_1] vector[N_1] z_1; // standardized group-level effects
}
transformed parameters {
vector[N_1] r_1_1; // actual group-level effects
real lprior = 0; // prior contributions to the log posterior
r_1_1 = (sd_1[1] * (z_1[1]));
lprior += normal_lpdf(Intercept | 0, 1);
lprior += student_t_lpdf(sigma | 3, 0, 2.5)
- 1 * student_t_lccdf(0 | 3, 0, 2.5);
lprior += normal_lpdf(sd_1 | 0, 1)
- 1 * normal_lccdf(0 | 0, 1);
}
model {
// likelihood including constants
if (!prior_only) {
// initialize linear predictor term
vector[N] mu = rep_vector(0.0, N);
mu += Intercept;
for (n in 1:N) {
// add more terms to the linear predictor
mu[n] += r_1_1[J_1[n]] * Z_1_1[n];
}
target += normal_lpdf(Y | mu, sqrt(square(sigma) + se2));
}
// priors including constants
target += lprior;
target += std_normal_lpdf(z_1[1]);
}
generated quantities {
// actual population-level intercept
real b_Intercept = Intercept;
}
Conclusion and Observations
There are two “take aways” from this post that we would like to emphasize.
-
brmsmakes it easy to specify Bayesian models that produce probability distributions that fully characterize estimated quantities. There is really no reason to settle for the frequentist point estimates that were the best that could be done before the availability of MCMC engines likestan. -
In Bayesian analysis, we use the structural assumption that unidentified population parameters generate individual outcomes that are used to create summaries of these individuals. This means knowing the individuals (or, in our case, sufficient statistics about the individuals) greatly influences our posterior estimates of plausible population parameters. With
brmswe are able to quickly infer the difference between treatment and control in a disciplined and documented fashion.
We would also like to point out that our simple example which is a typical meta-analysis project is a multi-level model built without having patient-level data. If we had the patient-level data from the Amplodipine study, then we might have considered a three-level nested model: subjects within treatment arms within studies. We avoided the first level by not having the data and the second level by directly modeling the difference between treatment arms.
-
See Details under
brmsformulain thebrmspackage PDF. -
See Details under
set_prioron page 211 of the package PDF.
Related



